- 2021-10-20 发布 |
- 37.5 KB |
- 8页


申明敬告: 本站不保证该用户上传的文档完整性,不预览、不比对内容而直接下载产生的反悔问题本站不予受理。
文档介绍
仪器分析 复习参考资料
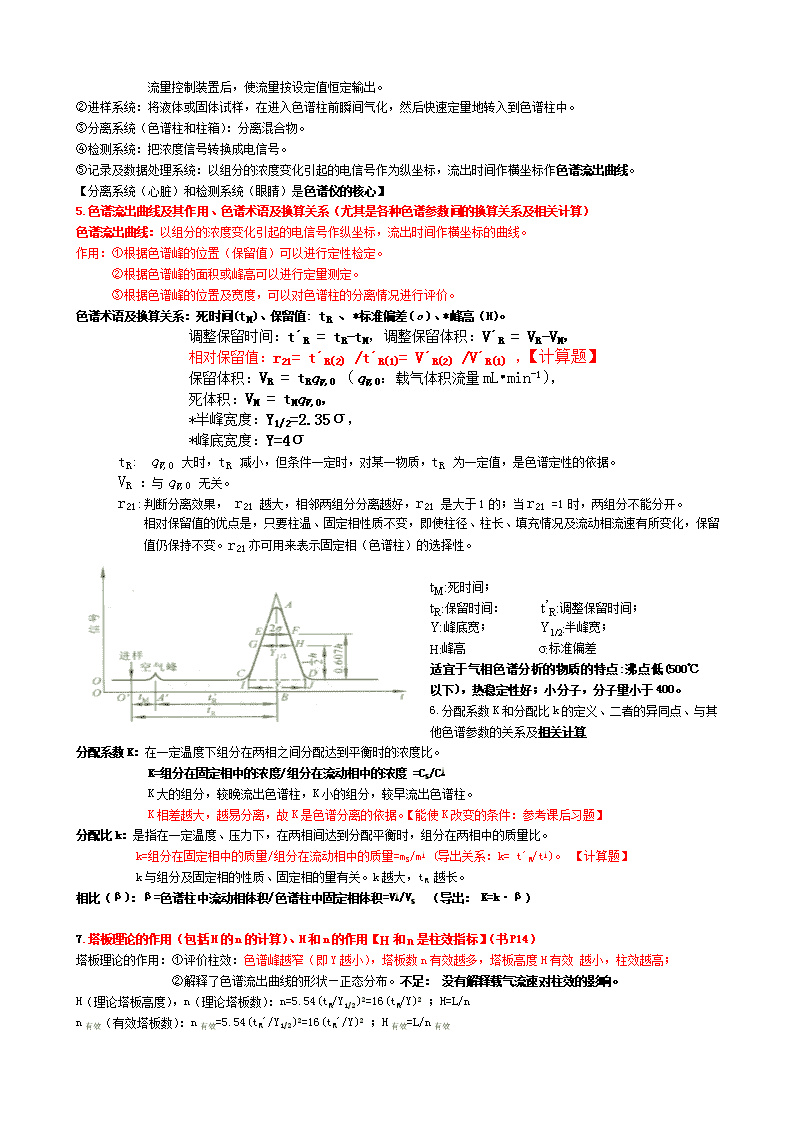
第1章 引言 1.什么是仪器分析法?主要类型有哪些? 仪器分析法:是以测量物质的物理性质为基础的分析方法。测定时常常需要借助比较复杂精密的仪器,故称“仪器分析法”。 类型: 光学分析法、电化学分析法(电位分析法、伏安分析法、库仑分析法) 色谱分析法(气相色谱法、液相色谱法及薄层色谱法) 其他方法(质谱法分析、热分析法) 第2章 气相色谱分析 1.色谱法分离原理:使混合物中各组分在两相间进行分配,流动相中的混合物经过固定相时,就会与固定相发生作用。由于各组分在性质和结构上的差异,与固定相发生作用的大小、强弱也有差异,因此在一推动作用下,不同组分在固定相中的滞留时间有长有短,从而按先后不同的次序从固定相中流出。即借助在两相间分配原理而使混合物中各组分分离。 固定相:管内保持不动、起分离作用的填充物(如CaCO3)。 流动相:流经固定相孔隙或表面的淋洗液(如石油醚)。 色谱法的分类(按两相状态) 按流动相状态:(1)气相色谱法GC(流动相是气态):按固定相——气—固色谱法、气—液色谱法; (2)液相色谱法LC(流动相是液态):按固定相——液—固色谱法、液—液色谱法 。 2.何为GC法,GC定性定量的依据、定量方法 GC法:即气相色谱法,是采用气体作为流动相的一种色谱法。 定性方法:1)保留值法 2)结合化学法 3)与质谱、红外光谱等一起联用 1)保留值法依据:在一定的色谱条件(固定相、操作条件)下,各种物质均有确定的保留值。据此,可与纯物质或文献对照保留值来定性。 定量的依据:在一定操作条件下,被测组分i的质量(mi)或浓度(Ci)与检测器的响应信号(峰面积Ai或峰高hi)成正比。表示为: mi=f´iAi ↔f´i= mi/Ai(f´为绝对质量校正因子)。 定量方法:1)归一化法 2)内标法 3)外标法。 1)归一化法的原理、使用条件 原理:试样质量为m,其中所含各组分质量为mi,则有: 其中i组分的质量分数 ωi 为: 使用条件:在测定条件下,试样中所有组分皆能流出色谱柱,并能在检测器产生信号,得到相应的色谱峰。 2)内标法及内标物具备的条件 内标物具备的条件:① 是试样中不存在的纯物质; ② 加入量应接近被测组分; ① 标物峰应在组分峰附近,或在几个组分峰之间,并与组分峰完全分离; ② 标物与被测组分的物理及物理化学性质(如挥发性、结构、极性、溶解性等)相近。 3)外标法:即标准曲线法或工作曲线法。数据处理 P268习题 配制标准系列,进样分别测峰面积A或峰高h,制作工作曲线 ,求得被测组分含量。 3.GC分离原理(包括GSC法和GLC法) 气-固色谱法(GSC):根据固定相对各组分的吸附能力的差异,对物质进行分离。固定相:固体吸附剂。 气-液色谱法(GLC):根据固定液对各组分的溶解能力的差异,对物质进行分离。固定相:固定液+担体。 气固色谱固定相常用的有活性炭、氧化铝、硅胶、分子筛、高分子多孔微球等。 分离特征是:按分子量由小到大的顺序流出柱子。 气-液色谱是利用固定液对混合物中不同组分具有不同溶解能力来实现分离的。 分离特征:遵循“相似相溶原理”。担体和固定液应具备的条件:见后面。 4.气相色谱仪的构造由五部分组成,各部分作用分别是: ① 载气系统:由气源输出的载气通过装有催化剂或分子的净化器,以除去水、氧等有害杂质,净化后的载气经稳压阀或自动流量控制装置后,使流量按设定值恒定输出。 ②进样系统:将液体或固体试样,在进入色谱柱前瞬间气化,然后快速定量地转入到色谱柱中。 ③分离系统(色谱柱和柱箱):分离混合物。 ④检测系统:把浓度信号转换成电信号。 ⑤记录及数据处理系统:以组分的浓度变化引起的电信号作为纵坐标,流出时间作横坐标作色谱流出曲线。 【分离系统(心脏)和检测系统(眼睛)是色谱仪的核心】 5.色谱流出曲线及其作用、色谱术语及换算关系(尤其是各种色谱参数间的换算关系及相关计算) 色谱流出曲线:以组分的浓度变化引起的电信号作纵坐标,流出时间作横坐标的曲线。 作用:①根据色谱峰的位置(保留值)可以进行定性检定。 ②根据色谱峰的面积或峰高可以进行定量测定。 ③根据色谱峰的位置及宽度,可以对色谱柱的分离情况进行评价。 色谱术语及换算关系:死时间(tM)、保留值: tR 、*标准偏差(σ)、*峰高(H)。 调整保留时间:t´R = tR-tM,调整保留体积:V´R = VR-VM, 相对保留值:r21= t´R(2) /t´R(1)= V´R(2) /V´R(1) ,【计算题】 保留体积:VR = tRqV,0 (qV,0:载气体积流量mL•min-1), 死体积:VM = tMqV,0, *半峰宽度:Y1/2=2.35σ, *峰底宽度:Y=4σ tR: qV,0 大时,tR 减小,但条件一定时,对某一物质,tR 为一定值,是色谱定性的依据。 VR :与qV,0 无关。 r21:判断分离效果, r21 越大,相邻两组分分离越好,r21 是大于1的;当r21 =1时,两组分不能分开。 相对保留值的优点是,只要柱温、固定相性质不变,即使柱径、柱长、填充情况及流动相流速有所变化,保留值仍保持不变。r21亦可用来表示固定相(色谱柱)的选择性。 tM:死时间; tR:保留时间: t’R:调整保留时间; Y:峰底宽; Y1/2:半峰宽; H:峰高 σ:标准偏差 适宜于气相色谱分析的物质的特点:沸点低(500℃以下),热稳定性好;小分子,分子量小于400。 6.分配系数K和分配比k的定义、二者的异同点、与其他色谱参数的关系及相关计算 分配系数K:在一定温度下组分在两相之间分配达到平衡时的浓度比。 K=组分在固定相中的浓度/组分在流动相中的浓度 =CS/CM K大的组分,较晚流出色谱柱,K小的组分,较早流出色谱柱。 K相差越大,越易分离,故K是色谱分离的依据。【能使K改变的条件:参考课后习题】 分配比k:是指在一定温度、压力下,在两相间达到分配平衡时,组分在两相中的质量比。 k=组分在固定相中的质量/组分在流动相中的质量=mS/mM (导出关系:k= t´R/tM)。 【计算题】 k与组分及固定相的性质、固定相的量有关。k越大,tR 越长。 相比(β):β=色谱柱中流动相体积/色谱柱中固定相体积=VM/VS (导出: K=k·β) 7.塔板理论的作用(包括H的n的计算)、H和n的作用【H和n是柱效指标】(书P14) 塔板理论的作用:①评价柱效:色谱峰越窄(即Y越小),塔板数n有效越多,塔板高度H有效 越小,柱效越高; ②解释了色谱流出曲线的形状—正态分布。不足: 没有解释载气流速对柱效的影响。 H(理论塔板高度),n(理论塔板数):n=5.54(tR/Y1/2)2=16(tR/Y)2 ;H=L/n n有效(有效塔板数):n有效=5.54(tR´/Y1/2)2=16(tR´/Y)2 ;H有效=L/n有效 8.速率理论方程的表达式、作用及其中各项的名称(包括u最佳、Hmin的计算) 从动力学观点出发,根据实验事实研究各种操作条件(载气的性质及流速、固定液膜厚度、柱温、载体颗粒大小、填充均匀度等)对塔板高度H(即柱效)的影响。影响H的因素为:涡流扩散项、分子扩散项、传质阻力项。并导出了速率理论方程。速率理论方程:H=A+B/u+Cu (H:塔板高度,u:载气线速度) (A:涡流扩散项,B:分子扩散项,C:传质阻力项) 作用:色谱分离条件选择的依据。 完全分离时:R=1.5 则n有效=16R2[a/(a-1)] 一般有效塔板数高度H有效=0.1cm 则L=n有效*H有效 由速率方程可知,改善柱效,应按以下原则考虑: 1.选择颗粒较小的均匀填料; 2.在不使固定液粘度增大的前提下,应在最低柱温下操作(有利于分离); 3.用最低实际浓度的固定液; 4.用较大摩尔质量的载气; 5.选择最佳载气流速。 9.R的含义、作用及计算 R的意义及作用:R是色谱柱的总分离效能指标。它表示色谱柱对相邻组分的分离效果。 R值越大,柱分离效能越高,分离效果越好。 相邻组分分离完全的标志:R=1.5。 当两组分分离效果差,且峰较宽时,可用半峰宽代替峰底宽计算分离度,此时表示为 10.四种检测器的载气、适用范围、注意事项及英文缩写 1)浓度型:①热导检测器(TCD)——H2 或He(热导系数大,灵敏度高) 检测原理:基于不同物质具有不同热导系数。 TCD对所有物质都有响应,属广谱型检测器。 ②电子捕获检测器(ECD)——N2(纯度大于99.99%,一定不含电负性物质) ECD是一种选择性检测器,适用于含氧、氮、硫、磷、卤素等电负性大的物质的测定。灵敏度高。 2)质量型:①火焰光度检测器(FPD)——N2 或H2 ; 对含S、P有机化合物具有高选择性和高灵敏度的质量型检测器。这种检测器可用于大气中痕量硫化物以及农副产品、水中的毫微克级有机磷和有机硫农药残留量的测定。 ②氢火焰离子化检测器(FID)——N2,适用于含碳有机物的测定。 注意事项:FID检测器的使用温度不能低于100℃,否则在空气中燃烧生成的水会在离子室凝聚成水,损坏离子室。 检测器的性能指标(5个) 1)灵敏度S:表示检测器响应信号对进样量的变化率。S值越大,表示检测器灵敏度越高。 2)检出限D:是指检测器恰能产生和噪声相鉴别的信号时,在单位体积或时间需向检测器进入的物质的质量(g)。 能与噪声鉴别开的最小信号:为噪声的3倍。 3)最小检出量:是指检测器能产生和噪声相鉴别的信号时进入色谱柱的最小物质量或最小浓度。 4)响应时间:是指从组分进入检测器到检测器有响应的时间。 5)线性范围:指检测器响应信号与试样量之间呈线性关系的范围。通常用浓度范围表示。 11.色谱分离操作条件及与柱效的关系,如何选择(书P21) 操作条件(载气的性质及流速、固定液膜厚度、柱温、载体颗粒大小、填充均匀度等)对塔板高度H(即柱效)的影响。 影响H的因素为:涡流扩散项、分子扩散项、传质阻力项。 1) 载气及其流速的选择 载气类型:气相色谱中所用的载气种类少,常用的有H2 、N2 、Ar 、He。选择何种载气,通常主要根据检测器。 TCD:H2 或He FID: N2 ECD:N2 (纯度大于99.99%) FPD:N2 或H2 载气流速(u): 在实际中,通常使流速稍高于最佳流速,以缩短分析时间。一般载气流速(柱内径3mm):N2:15~25mL•min-1 H2:30~40mL•min-1 结合速率理论方程H = A + B/u + Cu和塔板高度与载气流速的关系图,选择载气流速时: ★当载气流速较小时,分子扩散项( B/u )为色谱峰扩张的主要因素,应使用相对分子量较大的载气(N2,Ar)。 ★当载气流速较大时,传质阻力项( Cu )为色谱峰扩张的主要因素,应使用相对分子量较小的载气(H2,He)。 2)柱温的选择 柱温对分离的影响:柱温高,组分挥发快且靠拢,不易分离;柱温低,组分扩散慢,不能在两相中迅速达到分配平衡,峰拖尾扩张,柱效下降,且分析时间延长。 选择原则:在能使最难挥发的组分气化的 条件下,尽量使用较低的柱温;不能高于固定液的最高使用温度。 3)固定液的要求和用量 ① 挥发性小,在操作温度下有较低蒸汽压,以免流失。 ② 热稳定性好,在操作温度下不发生分解,且呈液态。 ③ 对试样各组分有适当的溶解能力,否则组分易被载气带走而起不到分配作用。 ④ 具有高的选择性,即对沸点相同或相近的不同物质有尽可能高的分离能力。 ⑤ 化学稳定性好,不与被测物质发生化学反应。 用量: 低固定液用量。液膜薄,柱效提高,缩短分析时间;固定液与担体的配比:5:100~25:100 4)担体的要求和粒度 ① 表面化学惰性,即无吸附或吸附很弱,不能与被测物质起化学反应。 ② 多孔,即表面积大,使固定液与试样的接触面较大。且表面和孔径分布均匀。 ③ 热稳定性好,有一定机械强度,不易碎。 ¢ 粒度:均匀细小。60~80目或80~100目。 5)进样时间和进样量: 进样时间:1s 进样量:液体0.1~5uL,气体0.1~10mL 6)汽化温度: 比柱温高30~70℃。 第3章 HPLC 高效液相色谱法主要类型:液-液色谱及化学键合相法、液-固色谱、离子对色谱法、离子交换色谱法和空间排阻色谱法等。 1.从分离原理、仪器构造及应用范围上简要比较气相色谱及液相色谱的异同点。 ①二者都是根据样品组分与流动相和固定相相互作用力的差别进行分离的。 ②从仪器构造上看,液相色谱需要增加高压泵以提高流动相的流动速度,克服阻力。同时液相色谱所采用的固定相种类要比气相色谱丰富的多,分离方式也比较多样。气相色谱的检测器主要采用热导检测器、氢焰检测器和火焰光度检测器等。而液相色谱则多使用紫外检测器、荧光检测器及电化学检测器等。但是二者均可与 MS等联用。 ③二者均具分离能力高、灵敏度高、分析速度快,操作方便等优点,但沸点太高的物质或热稳定性差的物质难以用气相色谱进行分析。而只要试样能够制成溶液,既可用于 HPLC分析,而不受沸点高、热稳定性差、相对分子量大的限制。 2.离子对色谱法、离子色谱法、离子交换色谱法的概念、适用特点(书P72-74)及英文缩写 离子对色谱法(IPC):是将一种(或数种)与溶质离子电荷相反的离子(称对离子或反离子)加到流动相或固定相中,使其与溶质离子结合形成疏水型/中性离子对化合物,从而控制溶质离子保留行为的一种色谱法。各种强极性的有机酸、有机碱特别是核酸、核苷、生物碱等的分离是离子对色谱的特点。 离子色谱法(IC):以分析水溶液中阴离子为主的液相色谱法。 离子交换色谱法(IEC):是利用离子交换原理和液相色谱技术的结合来测定溶液中阳离子和阴离子的一种分离分析方法。各种离子及在溶液中能够离解的物质均可实现分离,包括无机化合物、有机物及生物分子,如氨基酸、核酸及蛋白质等。 2.离子色谱法通常使用什么检测器:电导检测器。 3.什么是正相色谱法、反相色谱法、化学键合相色谱法 正相分配法:流动相的极性小于固定相的极性,适用于极性化合物的分离。其流出顺序是极性小的先流出,极性大的后流出。 反相分配法:流动相的极性大于固定相的极性,适用于非极性化合物的分离。其流出顺序与正相色谱恰好相反,极性大的先流出,极性小的后流出。 化学键合相色谱法:采用化学键合相为固定相的液相色谱法。能分离物质与液-固吸附色谱相同。 4.何谓梯度洗提?它与气相色谱中的程序升温有何异同之处? 在一个分析周期内,按一定程序不断改变流动相的组成或浓度配比,称为梯度洗提.是改进液相色谱分离的重要手段. 梯度洗提与气相色谱中的程序升温类似,但是前者连续改变的是流动相的极性、 pH或离子强度,而后者改变的温度.程序升温也是改进气相色谱分离的重要手段. 5液相色谱中影响色谱峰展宽的因素有哪些?与气相色谱相比较,有哪些主要不同之处? 液相色谱中引起色谱峰扩展的主要因素为涡流扩散、流动的流动相传质、滞留的流动相传质以及柱外效应。 在气相色谱中径向扩散往往比较显著,而液相色谱中径向扩散的影响较弱,往往可以忽略。另外,在液相色谱中还存在比较显著的滞留流动相传质及柱外效应。 第4章 电位分析法 1.电化学分析法:利用物质的电学及电化学性质进行分析的方法。亦称电分析化学法。 2.电位分析法的定量分析依据。 依据:能斯特方程。能斯特方程表示了电极电位与溶液中对应离子活度之间的定量关系。 电位分析法:是通过测定构成化学电池的电极的电极电位来确定被测物的含量的方法。 包括直接电位法(电位测定法)和电位滴定法。 直接电位法(电位测定法):根据电极电位测定值,直接求待测物的含量。 电位滴定法:测定原理同化学滴定法相同,只是以电极电位的突变代替了化学滴定中的指示剂颜色变化来确定滴定终点。 3.电位分析法测定溶液pH值的依据及方法(包括计算)、使用的电极及其结构特征 直接电位法测定溶液pH时,以玻璃电极为指示电极,甘汞电极(SCE)为参比电极,与待测液构成化学电池。 甘汞电极(SCE):电极电位恒定。甘汞电极电极反应:Hg2Cl2+2e- ↔ 2Hg+2Cl- 半电池的表示:Hg,Hg2Cl2(固)KCl 电极电位:(25℃) 甘汞电极的电极电位决定于电极内参比溶液KCI中的αCl- ,当αCl-一定,甘汞电极的电极电位恒定。 在其他温度下使用时,应对其电极电位进行校正。饱和甘汞电极(SCE)校正公式为: E=0.2438-7.6×10-4(t-25)(v) 玻璃电极:电极电位随被测溶液pH变化而变化。玻璃电极的主要组成部分是玻璃膜,膜内是H+浓度一定的内参比溶液,膜外是待测溶液。因膜内外H+浓度不相等,因此产生了膜电位,该膜电位与试液中H+浓度符合Nernst方程: ㏒ (㏒ -㏒ ) 是常数,令其为K,则有: log 在实际测定中,将玻璃电极与甘汞电极组成化学电池,通过测定该电池的电动势(E)来求得结果。 电位测定法的装置:①电位(pH)计; ②工作电池:由参比电极,指示电极,被测试液组成; ③磁力搅拌器(附磁力搅拌子)。 4.电位滴定法的原理、终点确定方法(包括三种方法的曲线特征及终点位置)。 指示电极:电极电位随被测离子浓度变化的电极。 电极 工作电池 参比电极:测定中电极电位为恒定值,不随被测离子浓度变化的电极。 电解液(待测液) 原理:随着滴定剂的加入,待测离子的浓度在不断变化,从而引起指示电极电位的变化,在化学计量点附近,电位的变化有一个突跃,根据此突跃确定滴定终点,由终点时消耗滴定剂的量以及滴定剂与被测物反应的化学计量关系,即可求得试液中待测组分的含量。 终点确定方法:【3个名称】 E/V曲线法:以滴定剂的体积(V)为横坐标,相应电动势读数(E)为纵坐标,绘制曲线。 曲线形状:梯形。 滴定终点:曲线转折点对应的V值。 ②ΔE/ΔV—V 曲线法(一级微商法):以滴定剂的体积(V)为横坐标,ΔE/ΔV为纵坐标,绘制曲线。 曲线形状:对称尖峰状。 滴定终点:尖峰点对应的V值。 ③Δ2E/Δ2V—V曲线法(二级微商法):以滴定剂的体积(V)为横坐标,Δ2E/Δ2V为纵坐标, 绘制曲线。 曲线形状:对称双曲线。 滴定终点:曲线上Δ2E/Δ2V=0对应的V值。 5、电位滴定法的优点(与普通滴定法比较) 1.灵敏、快速。 2.可用于浑浊、有色溶液的滴定。 3.可用于无合适指示剂或颜色变化不明显的反应。 4.可用于很弱的酸、碱或浓度很稀的溶液的滴定。 5.可用于混合物溶液的连续滴定。 6.易于实现自动化。 第8章 原子吸收光谱分析(AAS) 1.AAS的概念及基本原理(书P228-236) AAS:基于待测物元素基态原子蒸气对其特征谱线(通常是待测元素的特征谱线)的吸收作用来对物质进行定量分析的方法。 也称原子吸收光谱分析、原子吸收分光光度法、原子吸收法 2.与其它光谱分析法相比,AAS的干扰少,具有相对高选择性,为什么? 通常选择元素的共振线作为分析线,因为共振线作为分析线,测定灵敏度高。但在共振线附近若有干扰时,要选择干扰少、灵敏度稍低的非共振线为分析线。如Cu的共振线为324.754nm,而Eu为324.753nm,这时Eu对Cu有干扰,此时就应选Cu的次灵敏线324.4nm为分析线测定Cu ,从而避免Eu的干扰。 另外,在分析被测元素浓度较高试样时,可选用灵敏度较低的非共振线作为分析线。 3.玻兹曼方程对原子吸收光谱法定量依据的成立有作用P234 3.何为共振线?在AAS中,选择分析线的原则是什么? 当电子从基态跃迁到第一激发态时,与所吸收能量对应的光谱线叫共振吸收线;而由第一激发态跃迁回到基态时,与所释放能量对应的光谱线叫共振发射线。二者皆又称为共振线。 选择分析线要从两方面出发:灵敏度高;干扰少。 4.在AAS中,被测物质是何微粒形式?(找不到) 5.原子吸收分光光度计的基本部件组成?各部件的作用,常用何种光源?何种检测器?光源工作性能受什么影响? 原子吸收分光光度计基本部件有四部分:光源、原子化系统、光学系统、检测系统 光源→原子化器→分光系统→检测系统 作用①光源:发射待测元素基态原子的特征谱线。 ②原子化系统:将试样中的待测元素转变为基态的原子蒸气。 ③光学系统:包括两部分:照明系统(外光路系统)、分光系统(单色器)。 照明系统:作用是使光源发出的共振线能正确地通过被测试样的原子蒸气,并将其透射光投射到单色器上。 单色器:作用是可将被测元素的共振吸收线与邻近谱线分开。 ④检测系统:由检测器、放大器、对数转换器、显示装置等组成。在此主要介绍检测器、放大器。 检测器作用:将单色器分出的光信号转变为电信号。 放大器作用:将光电倍增管输出的电压信号进一步放大。 原子吸收分析中常用的光源是:空心阴级灯(HCL )使用前一般须预热10 ~ 30 min,检测器通常使用光电倍增管。 6.影响空心阴极灯发射特性的因素有哪些?关系如何? 空心阴极灯的发射特性取决于工作电流。灯电流过小,放电不稳定,光输出的强度弱;灯电流过大,发射谱线变宽,导致灵敏度下降,灯寿命缩短。(呼吸应急灯使用前要预热) 7.在火焰原子化中,影响火焰温度的因素是:火焰温度,所以,选择适宜的火焰条件是一项重要的工作。一般地,选 择的火焰温度应使待测元素恰能分解成基态自由原子为宜,不宜过高。 火焰温度的高低取决于火焰的组成,即燃气及助燃气的种类及流量。 8.火焰温度如何影响原子化效率?【简答】 若温度过高,会增加原子电离或激发,而使基态自由原子减少,原子化效率降低,导致分析灵敏度降低;若过低,不能使试样中化合物解离或解离率太小,同样降低原子化效率,灵敏度下降。 火焰温度与原子化效率间的关系可简单图示如下: 9.AAS法定量的基础、定量方法及相关计算(书P234-236) 解:在一定的浓度范围和一定的火焰宽度条件下,当采用锐线光源时,溶液的吸光度与待测元素浓度成正比关系,这就是原子吸收光谱定量分析的依据。 常用两种方法进行定量分析:【标准加入法的计算大题】 (1)标准曲线法:该方法简便、快速,但仅适用于组成简单的试样。 (2)标准加入法:本方法适用于试样的确切组分未知的情况。不适合于曲线斜率过小的情况。 10.AAS法适宜于常量分析还是微量分析?(找不到) 11. AAS法中的灵敏度、特征浓度、检出限 灵敏度(S):校正曲线的斜率。 特征浓度:是指能产生1%吸收(即吸光度值为0.0044)信号时所对应的溶液中被测元素的质量浓度 (μg•mL-1)或质量分数(μg•g-1 )。 检出限:指产生一个能确证在试样中存在某元素的分析信号所需要的该元素的最小含量。 12.AAS分析中,常见的干扰有哪些? 原子吸收光谱法的主要干扰有:光谱干扰(光源、原子化器)物理干扰、化学干扰(为主要来源)、有机溶剂 13.何为AAS法中的化学干扰?有哪些具体形式?如何消除?如何选择消电离剂? 化学干扰:是指在原子化过程中待测元素与其它组分发生化学反应等而引起的干扰。主要影响原子化效率。 分两种情况:①待测元素与其它组分发生化学反应,生成难挥发的化合物,导致原子化效率降低。 ②待测元素的电离。待测元素原子在火焰中失去电子而电离,使基态原子数减少,原子化效率降低。 消除方法:【3个名称】 入消电离剂:作用原理是在相同条件下消电离剂首先电离,消耗了能量并产生大量的电子,从而抑制、 减弱被测元素的电离。(加入的消电离剂跟被测元素关系: ) ②加入释放剂:作用是释放剂与干扰物质能生成比与被测元素更稳定或更难挥发的化合物,使被测元素释放出来。 ③加入保护剂:保护剂作用是它可与被测元素生成更稳定的配合物,抑制被测元素与干扰组份生成难离解的化合物。而保护剂与被测元素生成的化合物在原子化条件下又易于解离而原子化。保护剂一般是有机配合剂。例如,EDTA、8-羟基喹啉。 14.测定条件的选择:1. 分析线 2.灯电流 3. 火焰4.燃烧器高度5.狭缝宽度p260查看更多